
Dr. John Sayer’s Discovery of TULP3-related Ciliopathy, A Breakthrough in Gene Medicine
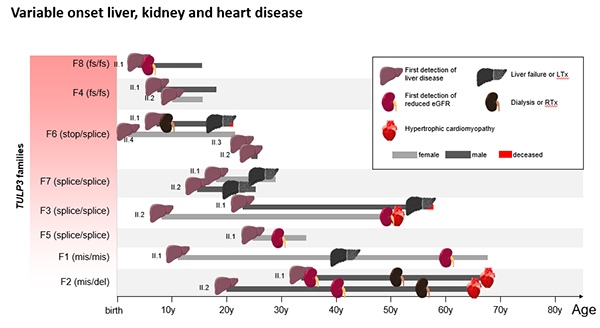
Professor John Sayer, Deputy Dean of Clinical Medicine at Newcastle University in the United Kingdom, a pioneer in kidney research and Genomics projects in the UK, has identified a faulty gene causing an inherited condition known as TULP3-related ciliopathy. This discovery is revolutionary in paving the way for proper diagnosis and tailored treatments in some patients who suffer from liver and kidney failures with unknown causes. In the recently published American Journal of Human Genetics, Dr. Sayer stated that his team reviewed clinical symptoms, collected liver biopsies and genetic sequencing from an array of patients, and identified 15 patients from eight families having this new disease. Among them, half of the patients had a liver or kidney transplant. The cause of these patients’ organ failures was unknown until this study.
We are very grateful that Dr. Sayer spent time explaining to us this important finding in the following interview. His discovery has tremendous implications for future gene therapy and organ transplants.
Q: Congratulations on your recent groundbreaking discovery that will help people suffering from unexplained liver and kidney issues. Please share with us your education and research background that led up to your discovery.
A: I am a clinician scientist and Professor of Renal Medicine at (Newcastle University). I have a long-standing interest in the genetics of kidney disease. I work closely with patients and families with rare and undiagnosed kidney diseases and strive to make a diagnosis for these individuals.
Q: Please share with us your research approach and your findings.
A: We have recently discovered a brand new inherited condition, called TULP3-related ciliopathy, that causes kidney and liver failure in children and adults. In this study, we reviewed clinical symptoms and genetic sequencing data from hundreds of patients in whom a genetic disease was suspected. We identified a total of 15 patients from eight families with mutations in TULP3 that accounted for their clinical symptoms.
Q: What is the significance of this discovery?
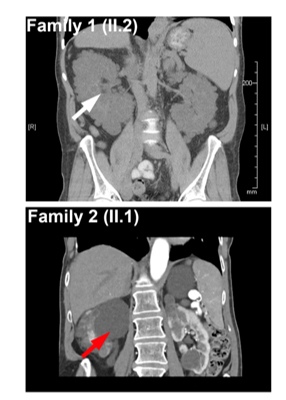
A: This study revealed that the children and adults had a brand-new genetic explanation for their disease. The gene, when faulty, leads to increased fibrosis in the liver and kidney and often results in organ damage that would require transplants. Urine samples from patients were used to grow patient cells and were investigated to determine the precise defect causing the disease.
Q: What is TULP3? How would you repair a defective TULP3 gene?
A: TULP3 is a protein that is critical for the function of the primary cilium, the antenna-like device present on most cells that allows the cells to sense their environment. Defects in cilia lead to downstream effects on the cell ultimately leading to tissue damage. The kidney and liver are particularly susceptible to defects in the cilia. There are several ways this condition may be treated and we are currently working on drugs that enable the cilia to work despite the TULP3 defect and gene editing approaches to repair the abnormal TULP3 gene.
Q: From your research, is the inherited condition, known as TULP3-related ciliopathy, found mainly in specific race/gender groups?
A: Our work looked globally for cases of TULP3 mutations. The cases we identified were from Europe and North America but we believe this condition affects all populations and that as a result of our work many more cases will be diagnosed.
Q: How would you incorporate this discovery into your ongoing treatments?
A: We are working with patient samples to use kidney cell lines to develop new treatments for TULP3-related ciliopathy. We have begun new collaborations across the globe to progress this work as rapidly as possible. The final common pathway of the TULP3 mutation is tissue fibrosis and this is an area of intense research.
Q: You are a member of the Epithelial Research Group at Newcastle University. What are the main tasks of your group and the goals it aims to achieve?
A: The Epithelial Research Group at Newcastle University is recognized as an international center for excellence in epithelial research. The mission is to understand epithelial processes at the cellular and molecular level and how their function and dysfunction relate to the whole organism in vivo. Within Newcastle University are a group of active researchers who can deploy a variety of methodologies ranging from sophisticated electrophysiological techniques through to whole organism physiology. This work was also performed from within Newcastle Centre for Rare Disease. Newcastle University has a stellar track record in rare disease research which has benefited from decades of collaboration between the University and Newcastle Upon Tyne Hospitals NHS Foundation Trust. Within this group, we collaborate across disciplines for patient-centered impact and innovation. We improve therapy options, outcomes, and quality of life for people with a rare disease.
Q: Moving forward, what are the next research projects that you would like to pursue?
A: Ongoing and future work will be to continue to perform gene discovery research in patients with rare diseases. The infrastructure at Newcastle University allows me to maximize my opportunities for cross-disease and cross-disciplinary research. We are exploring future therapies and are pursuing external collaborations to achieve this goal as efficiently as possible.

Dr. Sayer’s discovery provides new hope for many people who suffer from unexplained liver and kidney diseases. We look forward to more exciting news from Dr. Sayer and his team.